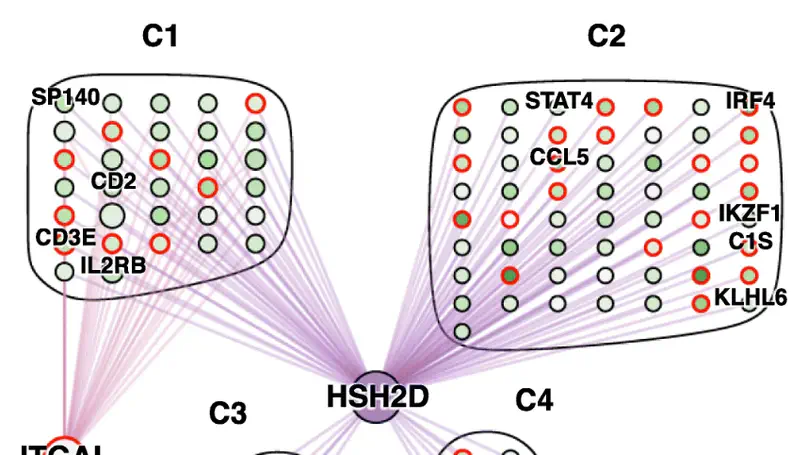
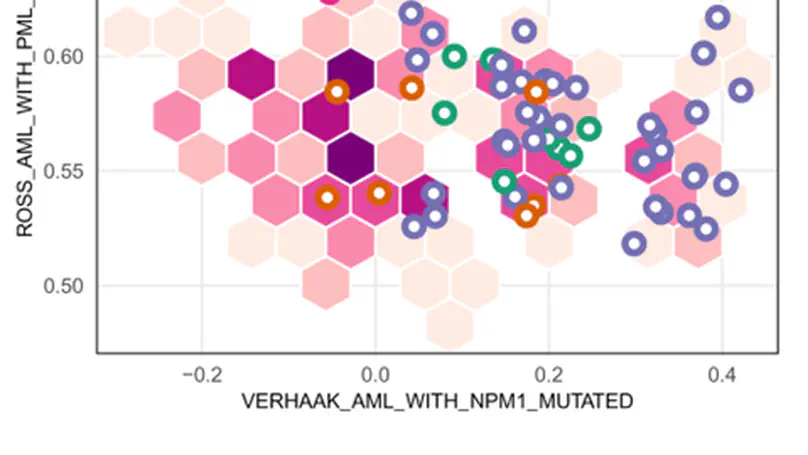
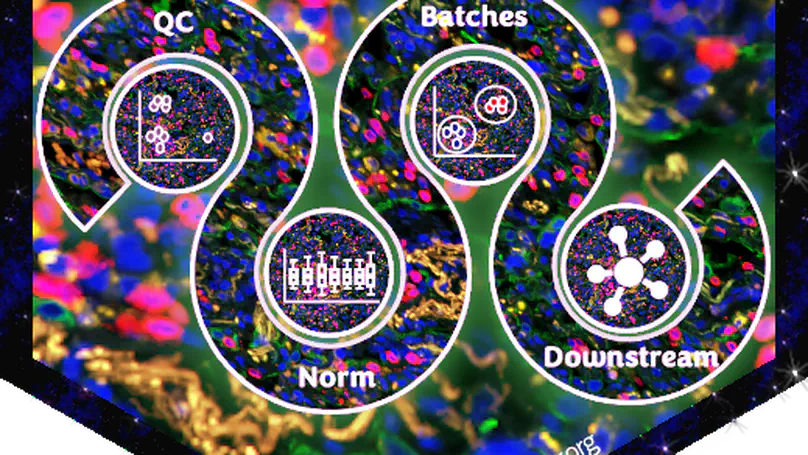
standR is an user-friendly R package providing functions to assist conducting good-practice analysis of Nanostring’s GeoMX DSP data. All functions in the package are built based on the SpatialExperiment object, allowing integration into various spatial transcriptomics-related packages from Bioconductor. standR allows data inspection, quality control, normalization, batch correction and evaluation with informative visualizations.