Differential co-expression-based detection of conditional relationships in transcriptional data: comparative analysis and application to breast cancer
 An immune-related regulatory module regulated specifically in estrogen receptor negative breast cancers
An immune-related regulatory module regulated specifically in estrogen receptor negative breast cancersAbstract
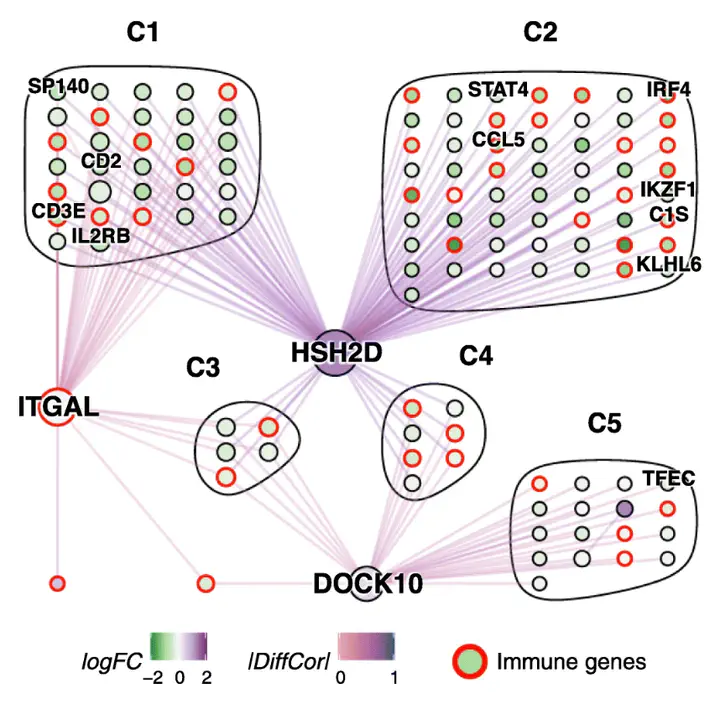
Background: Elucidation of regulatory networks, including identification of regulatory mechanisms specific to a given biological context, is a key aim in systems biology. This has motivated the move from co-expression to differential co-expression analysis and numerous methods have been developed subsequently to address this task; however, evaluation of methods and interpretation of the resulting networks has been hindered by the lack of known context-specific regulatory interactions. Results: In this study, we develop a simulator based on dynamical systems modelling capable of simulating differential co-expression patterns. With the simulator and an evaluation framework, we benchmark and characterise the performance of inference methods. Defining three different levels of “true” networks for each simulation, we show that accurate inference of causation is difficult for all methods, compared to inference of associations. We show that a z-score-based method has the best general performance. Further, analysis of simulation parameters reveals five network and simulation properties that explained the performance of methods. The evaluation framework and inference methods used in this study are available in the dcanr R/Bioconductor package. Conclusions: Our analysis of networks inferred from simulated data show that hub nodes are more likely to be differentially regulated targets than transcription factors. Based on this observation, we propose an interpretation of the inferred differential network that can reconstruct a putative causal network.
Dharmesh D Bhuva
Senior post-doctoral researcher at SAiGENCI
My research interests include cancer systems biology, spatial statistics and computational biology.