Biography
Dharmesh D Bhuva is an early career computational biology researcher (PhD in 2020) in the Davis laboratory at SAiGENCI. He has developed high quality bioinformatics software and methods that enable exploration of molecular phenotypes in cancer systems. He currently works on developing statistical and computational methods to analyse high throughput spatial transcriptomics datasets.
Download my resumé .
Interests
- Spatial Statistics
- Cancer Systems Biology
- Computational Biology
Education
PhD in Mathematics and Statistics, 2020
The University of Melbourne
MSc in Bioinformatics, 2015
The University of Melbourne
BSc in Computer Science, 2013
The University of Southampton
Experience
Senior Post-doctoral Researcher (Computational Systems Oncology)
Responsibilities include:
- Development of novel spatial statistics methods to analyse sub-cellular spatial ‘omics data.
- Development of machine learning pipelines to automate analysis of histopathology images.
- Student supervision
Post-doctoral Researcher (Bioinformatics and Biostatistics)
Responsibilities include:
- Analysis of spatial and single-cell transcriptomics to identify mechanisms of drug-induced phenotypic plasticity in cancer.
- Data analysis for commercial research collaboration between the CRC for Cancer Therapeutics and Pfizer Inc.
- Analysis of generic and drug specific regulatory mechanisms in biological systems using data integration approaches.
- Analysis of multi-omics data associated with drug response across a diverse set of biological models including cell lines and patients.
Projects
*
Recent & Upcoming Talks
*
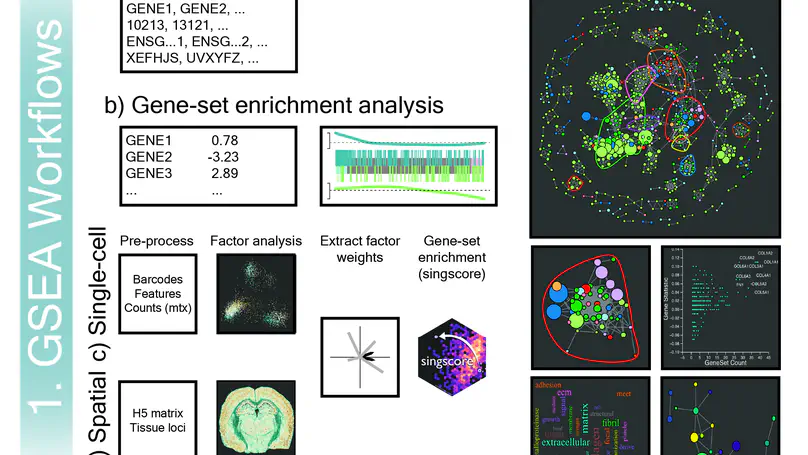
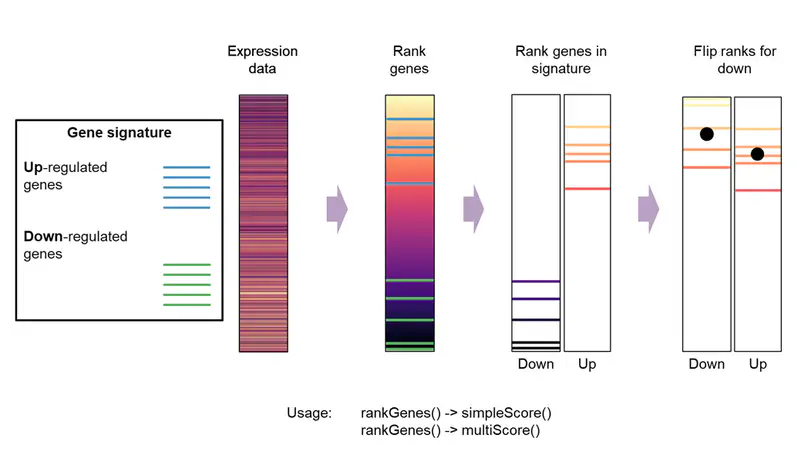
Recent Publications
(2023).
vissE.cloud: a webserver to visualise higher order molecular phenotypes from enrichment analysis.
(2022).
Transcriptomic profiling of cardiac tissues from SARS-CoV-2 patients identifies DNA damage.
Immunology.
(2022).
Profiling of lung SARS-CoV-2 and influenza virus infection dissects virus-specific host responses and gene signatures.
ERJ.
(2022).
Computational Screening of Anti-Cancer Drugs Identifies a New BRCA Independent Gene Expression Signature to Predict Breast Cancer Sensitivity to Cisplatin.
Cancers.
Recent softwares
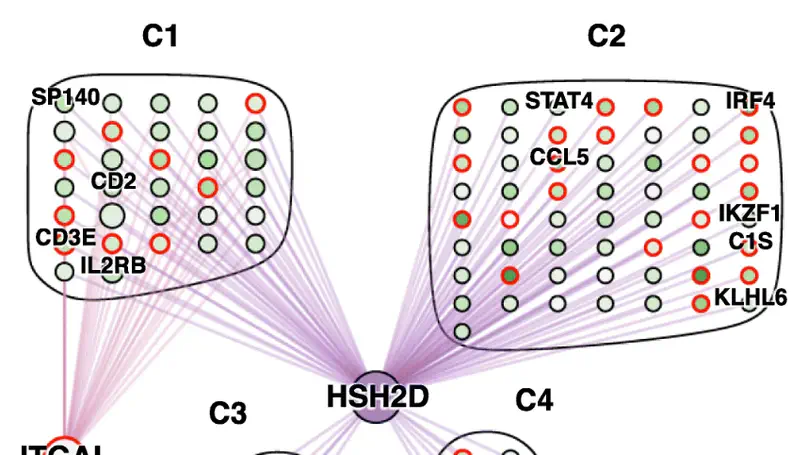

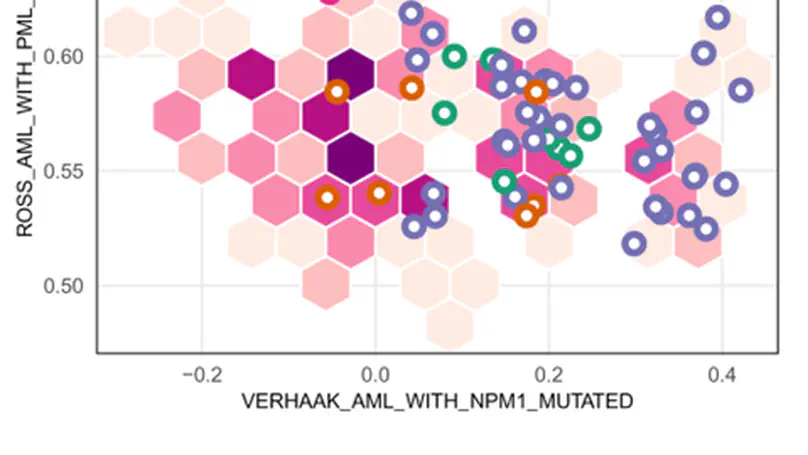

Contact
- test@example.org
- 888 888 88 88
- 450 Serra Mall, Stanford, CA 94305
- Enter Building 1 and take the stairs to Office 200 on Floor 2
-
Monday 10:00 to 13:00
Wednesday 09:00 to 10:00 - Book an appointment
- DM Me
- Zoom Me